Síndrome de angelman
El síndrome de Angelman (SA) es una afección genética que causa problemas con la forma en que se desarrollan el cuerpo y el cerebro de un niño. El síndrome está presente desde el nacimiento (congénito). Sin embargo, a menudo no se diagnostica hasta alrededor de los 6 a 12 meses de edad. Aquí es cuando los problemas de desarrollo se notan por primera vez en la mayoría de los casos.
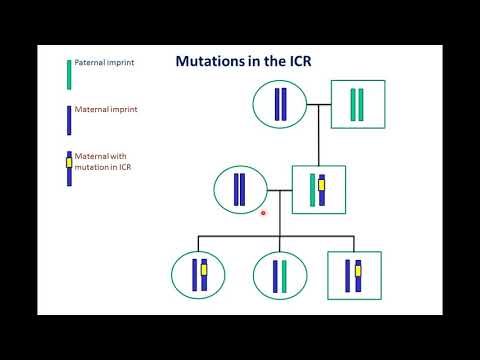
Esta condición involucra al gen UBE3A.
La mayoría de los genes vienen en pares. Los niños reciben uno de cada padre. En la mayoría de los casos, ambos genes están activos. Esto significa que las células utilizan la información de ambos genes. Con el UBE3A gen, ambos padres lo transmiten, pero solo el gen transmitido por la madre está activo.
El síndrome de Angelman ocurre con mayor frecuencia porque UBE3A transmitido de la madre no funciona como debería. En algunos casos, la EA se produce cuando dos copias de UBE3A El gen proviene del padre y ninguno proviene de la madre. Esto significa que ninguno de los genes está activo, porque ambos provienen del padre.
En recién nacidos y lactantes:
- Pérdida de tono muscular (flacidez)
- Problemas para alimentarse
- Acidez de estómago (reflujo ácido)
- Movimientos temblorosos de brazos y piernas
En niños pequeños y mayores:
- Caminar inestable o espasmódico
- Poco o nada de habla
- Personalidad alegre y excitable
- Reír y sonreír a menudo
- Color de cabello, piel y ojos claros en comparación con el resto de la familia
- Tamaño de cabeza pequeño en comparación con el cuerpo, parte posterior de la cabeza aplanada
- Discapacidad intelectual severa
- Convulsiones
- Movimiento excesivo de manos y extremidades.
- Problemas para dormir
- Empujar la lengua, babear
- Movimientos inusuales de masticación y boca
- Ojos cruzados
- Caminar con los brazos en alto y las manos agitando
La mayoría de los niños con este trastorno no muestran síntomas hasta alrededor de los 6 a 12 meses. Aquí es cuando los padres pueden notar un retraso en el desarrollo de su hijo, como no gatear o comenzar a hablar.
Los niños entre 2 y 5 años comienzan a mostrar síntomas como caminar entrecortado, personalidad feliz, reír a menudo, falta de habla y problemas intelectuales.
Las pruebas genéticas pueden diagnosticar el síndrome de Angelman. Estas pruebas buscan:
- Piezas faltantes de cromosomas
- Prueba de ADN para ver si las copias del gen de ambos padres están en un estado inactivo o activo
- Mutación genética en la copia del gen de la madre
Otras pruebas pueden incluir:
- Resonancia magnética cerebral
- Electroencefalograma
No existe cura para el síndrome de Angelman. El tratamiento ayuda a controlar los problemas de salud y desarrollo causados por la afección.
- Los medicamentos anticonvulsivos ayudan a controlar las convulsiones
- La terapia conductual ayuda a controlar la hiperactividad, los problemas de sueño y los problemas de desarrollo.
- La terapia ocupacional y del habla maneja los problemas del habla y enseña habilidades para la vida
- La fisioterapia ayuda con los problemas para caminar y moverse
Fundación del Síndrome de Angelman: www.angelman.org
AngelmanUK: www.angelmanuk.org
Las personas con EA viven cerca de una esperanza de vida normal. Muchos tienen amistades e interactúan socialmente. El tratamiento ayuda a mejorar la función. Las personas con AS no pueden vivir solas. Sin embargo, es posible que puedan aprender ciertas tareas y vivir con otras personas en un entorno supervisado.
Las complicaciones pueden incluir:
- Convulsiones severas
- Reflujo gastroesofágico (acidez de estómago)
- Escoliosis (columna vertebral curva)
- Lesión accidental debido a movimientos incontrolados
Llame a su proveedor de atención médica si su hijo presenta síntomas de esta afección.
No hay forma de prevenir el síndrome de Angelman. Si tiene un hijo con AS o antecedentes familiares de la afección, es posible que desee hablar con su proveedor antes de quedar embarazada.
Dagli AI, Mueller J, Williams CA. Síndrome de Angelman. GeneReviews. Seattle, WA: Universidad de Washington; 2015: 5. PMID: 20301323 www.ncbi.nlm.nih.gov/pubmed/20301323. Actualizado el 27 de diciembre de 2017. Consultado el 1 de agosto de 2019.
Kumar V, Abbas AK, Aster JC. Enfermedades genéticas y pediátricas. En: Kumar V, Abbas AK, Aster JC, eds. Patología básica de Robbins. 10ª ed. Filadelfia, PA: Elsevier; 2018: capítulo 7.
Madan-Khetarpal S, Arnold G. Trastornos genéticos y condiciones dismórficas. En: Zitelli BJ, McIntire SC, Nowalk AJ, eds. Atlas de diagnóstico físico pediátrico de Zitelli y Davis. 7ª ed. Filadelfia, PA: Elsevier; 2018: capítulo 1.
Nussbaum RL, McInnes RR, Willard HF. La base cromosómica y genómica de la enfermedad: trastornos de los autosomas y cromosomas sexuales. En: Nussbaum RL, McInnes RR, Willard HF, eds. Thompson & Thompson Genetics in Medicine. 8ª ed. Filadelfia, PA: Elsevier; 2016: capítulo 6.
Publicaciones Interesantes

34 maneras de despertarse sintiéndose renovado y listo para comenzar
