Cómo reconocer el síndrome de Rett

Contenido
- Características del síndrome de Rett
- Cómo se hace el diagnóstico
- Expectativa de vida
- ¿Qué causa el síndrome de Rett?
- Tratamiento para el síndrome de Rett
El síndrome de Rett, también llamado hiperamonemia cerebroatrófica, es una enfermedad genética rara que afecta al sistema nervioso y afecta casi exclusivamente a las niñas.
Los niños con síndrome de Rett dejan de jugar, se aíslan y pierden las habilidades aprendidas, como caminar, hablar o incluso mover las manos, dando lugar a movimientos involuntarios de las manos que son característicos de la enfermedad.
El síndrome de Rett no tiene cura, pero se puede controlar con el uso de medicamentos que reducen los ataques epilépticos, la espasticidad y la respiración, por ejemplo. Pero la fisioterapia y la estimulación psicomotora son de gran ayuda, y deben realizarse preferentemente a diario.
Características del síndrome de Rett
A pesar de que los síntomas que más llaman la atención de los padres aparecen recién después de los 6 meses de vida, el bebé con síndrome de Rett presenta hipotonía, y puede ser visto por padres y familiares, como un bebé muy 'bueno' y fácil de cuidar.
Este síndrome se desarrolla en 4 fases y en ocasiones el diagnóstico solo llega alrededor del año de edad, o más tarde, dependiendo de los signos que presente cada niño.
Primera fase, ocurre entre los 6 y los 18 meses de vida, y existen:
- Detener el desarrollo del niño;
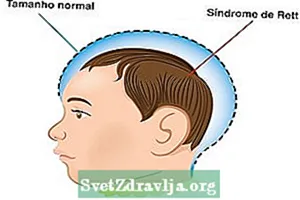
- La circunferencia de la cabeza no sigue la curva de crecimiento normal;
- Disminución del interés por otras personas o niños, con tendencia a aislarse.
Segunda fase, ocurre a partir de los 3 años y puede durar semanas o meses:
- El niño llora mucho, incluso sin motivo aparente;
- El niño siempre permanece irritado;
- Aparecen movimientos repetitivos de la mano;
- Aparecen cambios respiratorios, con la respiración detenida durante el día, un momento de aumento de la frecuencia respiratoria;
- Convulsiones convulsivas y ataques de epilepsia a lo largo del día;
- Los trastornos del sueño pueden ser comunes;
- El niño que ya habló, puede dejar de hablar por completo.


Tercera fase, que ocurre hace alrededor de 2 y 10 años:
- Puede haber alguna mejora en los síntomas presentados hasta ahora y el niño puede volver a mostrar interés en los demás;
- La dificultad para mover el tronco es evidente, hay dificultad para pararse;
- Puede haber espasticidad;
- Se desarrolla escoliosis que altera la función pulmonar;
- Es común rechinar los dientes durante el sueño;
- La alimentación puede ser normal y el peso del niño también suele ser normal, con un ligero aumento de peso;
- El niño puede perder el aliento, tragar aire y tener demasiada saliva.
Cuarta fase, que ocurre alrededor de los 10 años de edad:
- Pérdida de movimiento poco a poco y empeoramiento de la escoliosis;
- La deficiencia mental se vuelve severa;
- Los niños que podían caminar pierden esta capacidad y necesitan una silla de ruedas.
Los niños que pueden aprender a caminar todavía tienen algunas dificultades para moverse y generalmente andan de puntillas o dan los primeros pasos hacia atrás. Además, es posible que no puedan llegar a ninguna parte y su caminar parece no tener sentido porque ella no camina para encontrarse con otra persona o para recoger juguetes, por ejemplo.
Cómo se hace el diagnóstico
?????? El diagnóstico lo realiza el neuropediatra quien analizará en detalle a cada niño, de acuerdo con los signos presentados. Para el diagnóstico, se deben observar al menos las siguientes características:
- Desarrollo aparentemente normal hasta los 5 meses de vida;
- Tamaño normal de la cabeza al nacer, pero no sigue la medida ideal después de 5 meses de vida;
- Pérdida de la capacidad de mover las manos normalmente alrededor de los 24 y 30 meses, dando lugar a movimientos incontrolados como torcer o llevarse las manos a la boca;
- El niño deja de interactuar con otras personas al comienzo de estos síntomas;
- Falta de coordinación de los movimientos del tronco y caminar descoordinado;
- El niño no habla, no puede expresarse cuando quiere algo y no comprende cuando le hablamos;
- Retraso severo en el desarrollo, con sentarse, gatear, hablar y caminar mucho más tarde de lo esperado.
Otra forma más confiable de saber si este síndrome es realmente es mediante una prueba genética porque alrededor del 80% de los niños con síndrome de Rett clásico tienen mutaciones en el gen MECP2. El SUS no puede realizar este examen, pero los planes de salud privados no pueden negarlo, y si esto sucede, debe presentar una demanda.
Expectativa de vida
Los niños diagnosticados con el síndrome de Rett pueden vivir mucho tiempo, más allá de los 35 años, pero pueden sufrir muerte súbita mientras duermen, cuando aún son bebés. Algunas situaciones que favorecen complicaciones graves que pueden ser fatales incluyen la presencia de infecciones, las enfermedades respiratorias que se desarrollan debido a la escoliosis y la mala expansión pulmonar.
El niño puede asistir a la escuela y puede aprender ciertas cosas, pero lo ideal es que se integre a la educación especial, donde su presencia no atraerá mucha atención, lo que podría perjudicar su interacción con los demás.
¿Qué causa el síndrome de Rett?
El síndrome de Rett es una enfermedad genética y, por lo general, los niños afectados son los únicos en la familia, a menos que tengan un hermano gemelo, que probablemente tendrá la misma enfermedad. Esta enfermedad no está asociada con ninguna acción que hayan tomado los padres y, por lo tanto, los padres no deben sentirse culpables.
Tratamiento para el síndrome de Rett
El tratamiento debe ser realizado por el pediatra hasta que el niño tenga 18 años y, posteriormente, debe ser seguido por un médico de cabecera o neurólogo.
Las consultas deben realizarse cada 6 meses y en ellas se pueden observar signos vitales, talla, peso, corrección de medicación, valoración del desarrollo infantil, alteraciones cutáneas como la presencia de heridas por decúbito, que son las escaras que pueden infectarse, aumentando la riesgo de muerte. Otros aspectos que pueden ser importantes son la evaluación del desarrollo y del sistema respiratorio y circulatorio.
La fisioterapia debe realizarse durante toda la vida de la persona con síndrome de Rett y es útil para mejorar el tono, la postura, la respiración y se pueden utilizar técnicas como Bobath para ayudar al desarrollo del niño.
Las sesiones de estimulación psicomotora se pueden realizar aproximadamente 3 veces por semana y pueden ayudar con el desarrollo motor, disminuir la gravedad de la escoliosis, el control de la baba y la interacción social, por ejemplo. El terapeuta puede indicar algunos ejercicios que los padres pueden realizar en casa para que el estímulo neurológico y motor se realice a diario.
Tener a una persona con síndrome de Rett en casa es una tarea ardua y difícil. Los padres pueden sentirse muy agotados emocionalmente, por lo que se les puede recomendar que los sigan psicólogos que puedan ayudarlos a lidiar con sus emociones.
Popular En El Sitio

La relación entre el TDAH y el autismo
